BLAST & Databases
Identification of organism using standard databases:
What is a Database?
A database for DNA, RNA, and proteins is a specialized repository that stores biological information related to these essential molecules. These databases organize and provide access to sequences, structures, functions, and interactions of nucleic acids and proteins. Key features include searchable sequences, annotations about gene functions, variations, and evolutionary data. Prominent examples include GenBank for DNA sequences, UniProt for protein sequences and functions, and RNAcentral for RNA sequences. These resources are invaluable for researcher’s studying genetics, molecular biology, and bioinformatics, as they facilitate data analysis, comparison, and visualization in their exploration of the molecular basis of life. Along with the above-mentioned database, there are several specialized databases which can be used for specific target markers such as UNITE for ITS sequences and SILVA for 16S rRNA, etc.
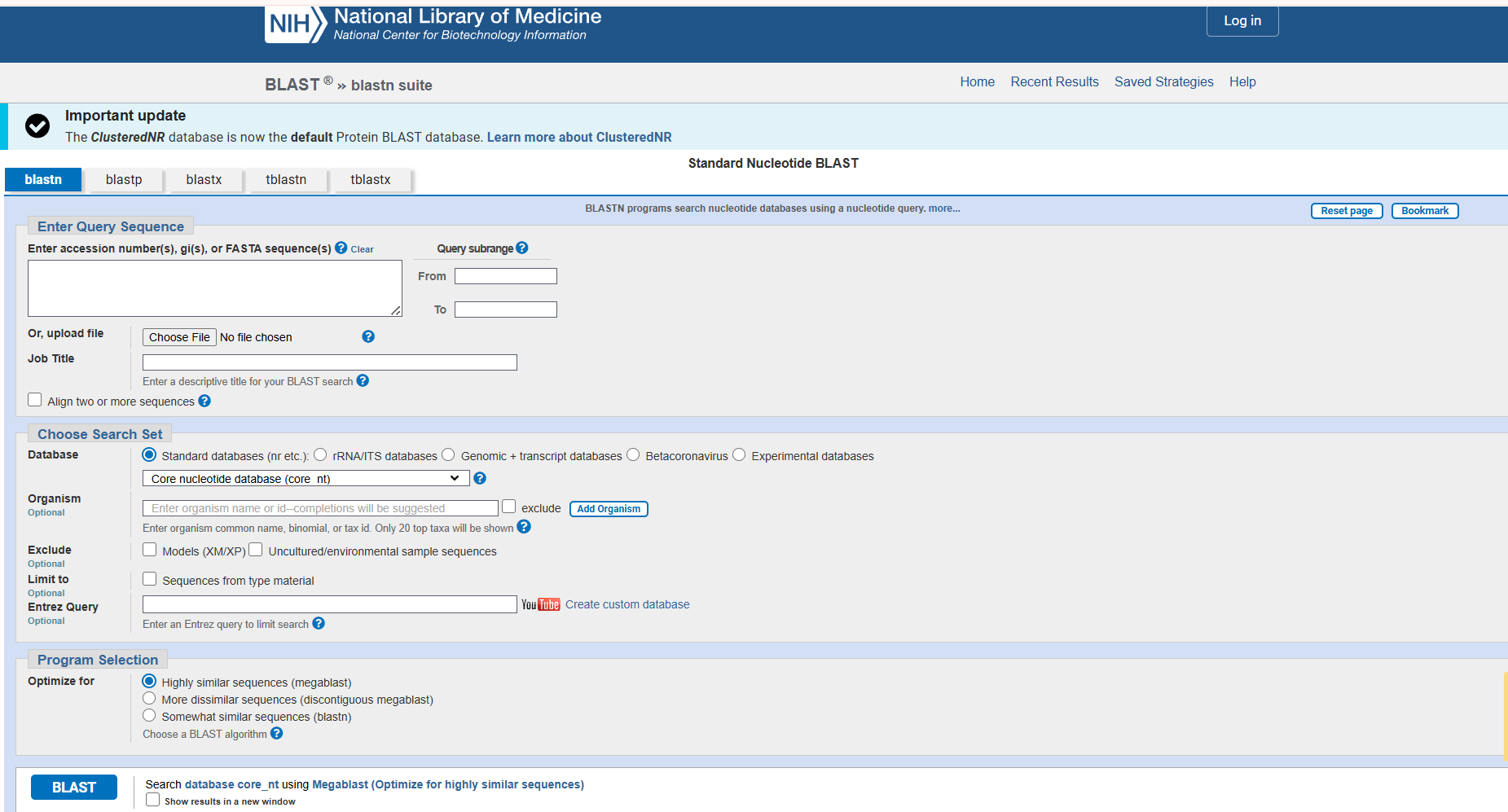
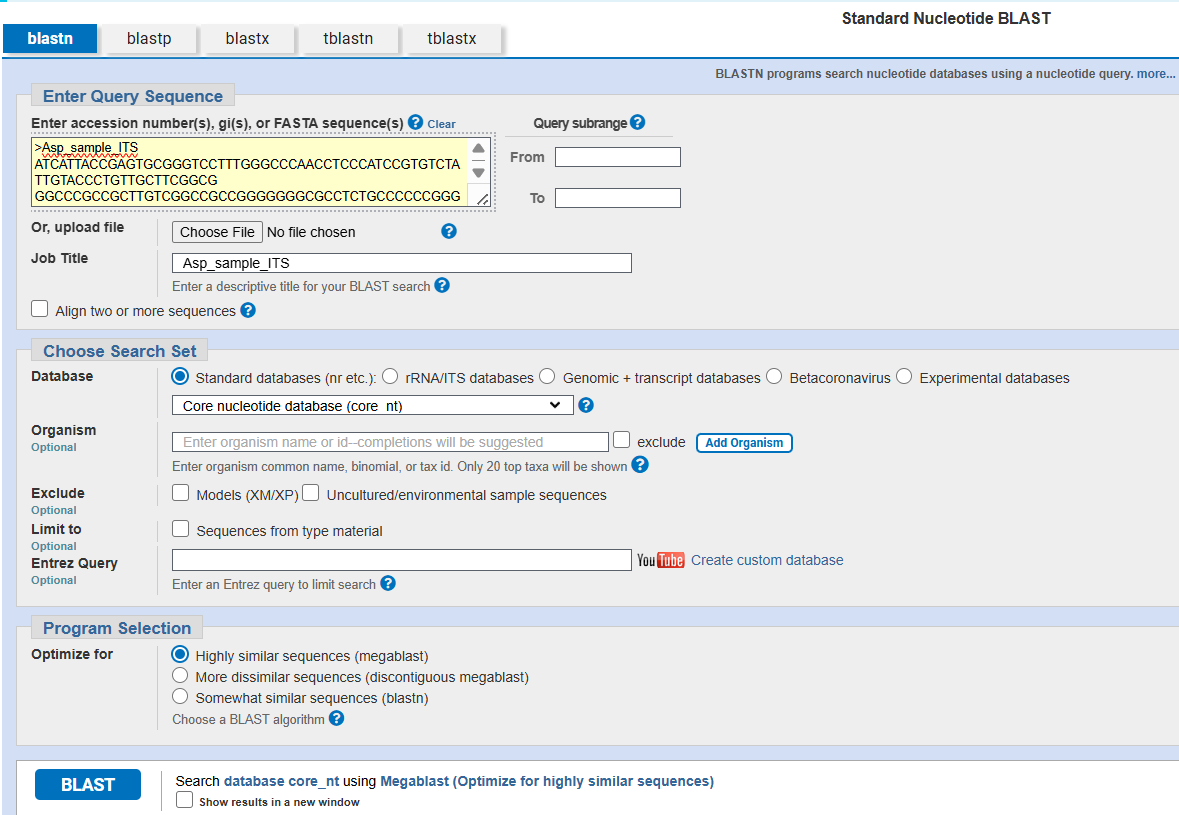
The correct identification is an important step to accurately identify the organisms before using them in any further application. The above consensus sequences provided for practice can be used for the dentification purpose using the BLAST tool of GenBank (NCBI). The following steps can be used to correctly identify / match the organisms using NCBI database (Fig. 10, and 15).
Steps to be followed (Fig. 10 to 15):
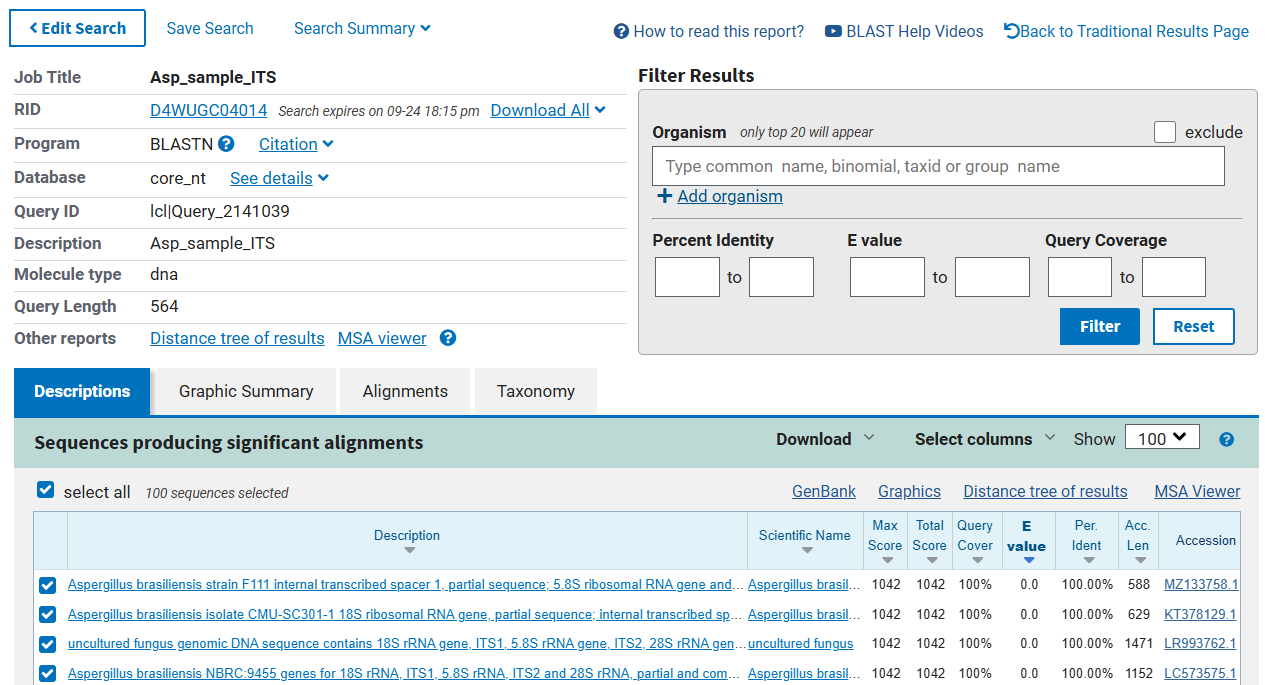
Interpretation of the BLAST result:
The Basic Local Alignment Search Tool (BLAST) is a powerful bioinformatics tool used to compare nucleotide or protein sequences against a database. When interpreting a BLAST result, you should focus on several key components:
1. Query Sequence
This is the sequence you submitted for comparison. Understanding this sequence is crucial for interpreting the results accurately.
2. Subject Sequences
These are the sequences from the database that showed similarities to your query. Each hit represents a potential match.
3. E-value (Expect value)
The E-value indicates the number of hits one might expect to see by chance when searching a database of a particular size. A lower E-value (typically less than 0.01 or close to 0) suggests a more statistically significant match.
4. Score
This reflects the quality of the alignment, considering both matches and mismatches. Higher scores indicate better alignments.
5. % Identities
This section shows how many of the residues in the query match the subject sequence (identities) and how many are similar (positives). These values help assess the degree of similarity. A higher % identity suggests a closer evolutionary relationship and potentially shared functions.
6. Alignment
This is the actual alignment of the sequences, highlighting matches, mismatches, and gaps. Visualizing the alignment can help you understand the regions of conservation or variability.
7. Organism Information
Knowing the organism from which the matched sequences are derived can provide context and relevance, especially if you’re studying a specific biological question.
8. Accession Numbers
These are unique identifiers for the subject sequences in the database, allowing further investigation into specific sequences or publications related to them.
As per the blast hit, the BLAST run will fetch several details (mentioned above) from the database, and it will show details as mentioned in figure 15.
| Percent Identity | Interpretation | Significance |
|---|---|---|
| > 97% | Strong conservation | Indicates strong conservation, often same genus and species or gene families (for microbes). Please note that in case of higher eukaryotes the % similarity alone cannot define the identity. |
| 90% – 97% | Strong conservation | Indicates strong conservation, often seen in closely related genus/ species or gene families. But the species-level identity will not be reliable. |
| 70% – 90% | Strong similarity | May still represent strong similarity and could suggest functional conservation between genus/ families. |
| 50% – 70% | Moderate similarity | Moderate similarity that could indicate shared ancestry but may also reflect divergent evolution. |
| < 50% | Low similarity | Often indicates that the sequences are distantly related. |